Full Guide
Bactopia is a complete pipeline for the analysis of bacterial genomes, which includes more than 150 bioinformatics tools. In this section, each step of the pipeline will be described in detail. This includes the input data, output data, and the parameters for each step.
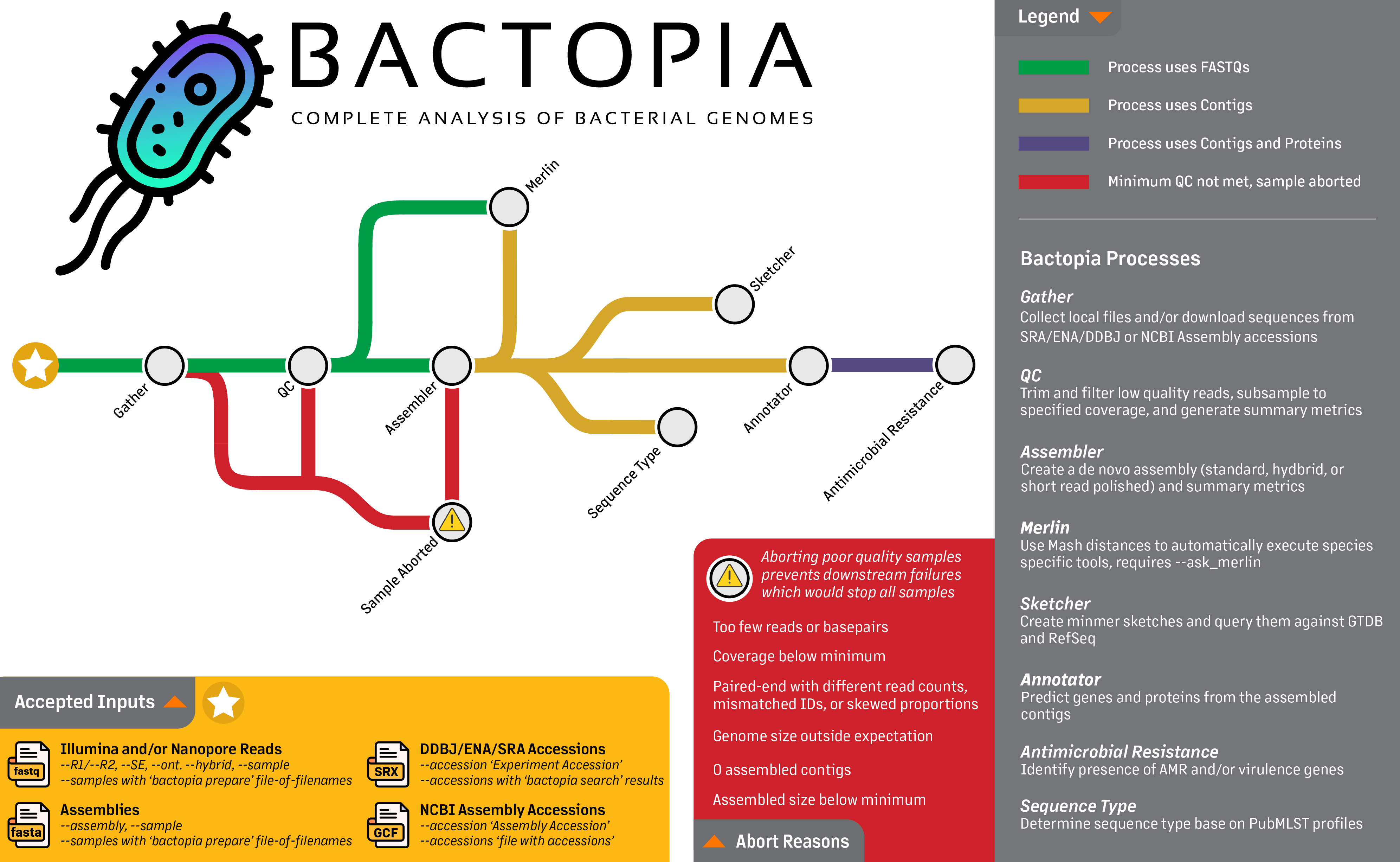
Looking at the workflow overview below, it might not look like much is happening, but I can assure you that a lot is going on. The workflow is broken down into 8 steps, which are:
- Gather - Collect all the data in one place
- QC - Quality control of the data
- Assembler - Assemble the reads into contigs
- Annotator - Annotate the contigs
- Sketcher - Create a sketch of the contigs, and query databases
- Sequence Typing - Determine the sequence type of the contigs
- Antibiotic Resistance - Determine the antibiotic resistance of the contigs and proteins
- Merlin - Automatically run species-specific tools based on distance
This guide is meant to be extensive, so it will be very detailed. If you are looking for a guide to get started quickly, please check out the Beginner's Guide.
Otherwise, let's get started!
Step 1 - Gather¶
The main purpose of the gather step is to get all the samples into a single place. This
includes downloading samples from ENA/SRA or NCBI Assembly. The tools used are:
| Tool | Description |
|---|---|
| art | For simulating error-free reads for an input assembly |
| fastq-dl | Downloading FASTQ files from ENA/SRA |
| ncbi-genome-download | Downloading FASTA files from NCBI Assembly |
This gather step also does basic QC checks to help prevent downstream failures.
Outputs¶
Merged Results¶
Below are results that are concatenated into a single file.
| Filename | Description |
|---|---|
| meta.tsv | A tab-delimited file with bactopia metadata for all samples |
gather¶
Below is a description of the per-sample results from the gather subworkflow.
| Filename | Description |
|---|---|
| -meta.tsv | A tab-delimited file with bactopia metadata for each sample |
Failed Quality Checks¶
Built into Bactopia are few basic quality checks to help prevent downstream failures. If a sample fails one of these checks, it will be excluded from further analysis. By excluding these samples, complete pipeline failures are prevented.
| Filename | Description |
|---|---|
| -gzip-error.txt | Sample failed Gzip checks and excluded from further analysis |
| -low-basepair-proportion-error.txt | Sample failed basepair proportion checks and excluded from further analysis |
| -low-read-count-error.txt | Sample failed read count checks and excluded from further analysis |
| -low-sequence-depth-error.txt | Sample failed sequenced basepair checks and excluded from further analysis |
Poor samples are excluded to prevent downstream failures
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: Input FASTQ(s) failed Gzip checks
If input FASTQ(s) fail to pass Gzip test, the sample will be excluded from further analysis.
Example Text from <SAMPLE_NAME>-gzip-error.txt
<SAMPLE_NAME> FASTQs failed Gzip tests. Please check the input FASTQs. Further
analysis is discontinued.
Example Error: Input FASTQs have disproportionate number of reads
If input FASTQ(s) for a sample have disproportionately different number of reads
between the two pairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_proportion parameter.
Example Text from <SAMPLE_NAME>-low-basepair-proportion-error.txt
<SAMPLE_NAME> FASTQs failed to meet the minimum shared basepairs (X``). They
sharedYbasepairs, with R1 havingAbp and R2 havingB` bp. Further
analysis is discontinued.
Example Error: Input FASTQ(s) has too few reads
If input FASTQ(s) for a sample have less than the minimum required reads, the
sample will be excluded from further analysis. You can adjust this minimum read
count using the --min_reads parameter.
Example Text from <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Example Error: Input FASTQ(s) has too little sequenced basepairs
If input FASTQ(s) for a sample fails to meet the minimum number of sequenced
basepairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_basepairs parameter.
Example Text from <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
Parameters¶
Required¶
The following parameters are how you will provide either local or remote samples to be processed by Bactopia.
| Parameter | Description |
|---|---|
--samples |
A FOFN (via bactopia prepare) with sample names and paths to FASTQ/FASTAs to process Type: string |
--r1 |
First set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r2 and --sample) Type: string |
--r2 |
Second set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r1 and --sample) Type: string |
--se |
Compressed (gzip) Illumina single-end FASTQ reads (requires --sample) Type: string |
--ont |
Compressed (gzip) Oxford Nanopore FASTQ reads (requires --sample) Type: string |
--hybrid |
Create hybrid assembly using Unicycler. (requires --r1, --r2, --ont and --sample) Type: boolean |
--short_polish |
Create hybrid assembly from long-read assembly and short read polishing. (requires --r1, --r2, --ont and --sample) Type: boolean |
--sample |
Sample name to use for the input sequences Type: string |
--accessions |
A file containing ENA/SRA Experiment accessions or NCBI Assembly accessions to processed Type: string |
--accession |
Sample name to use for the input sequences Type: string |
--assembly |
A assembled genome in compressed FASTA format. (requires --sample) Type: string |
--check_samples |
Validate the input FOFN provided by --samples Type: boolean |
Dataset¶
Define where the pipeline should find input data and save output data.
| Parameter | Description |
|---|---|
--species |
Name of species for species-specific dataset to use Type: string |
--ask_merlin |
Ask Merlin to execute species specific Bactopia tools based on Mash distances Type: boolean |
--coverage |
Reduce samples to a given coverage, requires a genome size Type: integer, Default: 100 |
--genome_size |
Expected genome size (bp) for all samples, required for read error correction and read subsampling Type: string, Default: 0 |
--use_bakta |
Use Bakta for annotation, instead of Prokka Type: boolean |
Citations¶
If you use Bactopia and gather results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
ART
Huang W, Li L, Myers JR, Marth GT ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 (2012) -
fastq-dl
Petit III RA fastq-dl: Download FASTQ files from SRA or ENA repositories. (GitHub) -
fastq-scan
Petit III RA fastq-scan: generate summary statistics of input FASTQ sequences. (GitHub) -
ncbi-genome-download
Blin K ncbi-genome-download: Scripts to download genomes from the NCBI FTP servers (GitHub) -
Pigz
Adler M. pigz: A parallel implementation of gzip for modern multi-processor, multi-core machines. Jet Propulsion Laboratory (2015)
Step 2 - QC¶
The qc module uses a variety of tools to perform quality control on Illumina and
Oxford Nanopore reads. The tools used are:
| Tool | Technology | Description |
|---|---|---|
| bbtools | Illumina | A suite of tools for manipulating reads |
| fastp | Illumina | A tool designed to provide fast all-in-one preprocessing for FastQ files |
| fastqc | Illumina | A quality control tool for high throughput sequence data |
| fastq_scan | Nanopore | A tool for quickly scanning FASTQ files |
| lighter | Illumina | A tool for correcting sequencing errors in Illumina reads |
| NanoPlot | Nanopore | A tool for plotting long read sequencing data |
| nanoq | Nanopore | A tool for calculating quality metrics for Oxford Nanopore reads |
| porechop | Nanopore | A tool for removing adapters from Oxford Nanopore reads |
| rasusa | Nanopore | Randomly subsample sequencing reads to a specified coverage |
Similar to the gather step, the qc step will also stop samples that fail to meet
basic QC checks from continuing downstream.
Outputs¶
Quality Control¶
Below is a description of the per-sample results from qc subworkflow.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.fastq.gz | A gzipped FASTQ file containing the cleaned Illumina single-end, or Oxford Nanopore reads |
| <SAMPLE_NAME>_R{1|2}.fastq.gz | A gzipped FASTQ file containing the cleaned Illumina paired-end reads |
| <SAMPLE_NAME>-{final|original}.json | A JSON file containing the QC results generated by fastq-scan |
| <SAMPLE_NAME>-{final|original}_fastqc.html | (Illumina Only) A HTML report of the QC results generated by fastqc |
| <SAMPLE_NAME>-{final|original}_fastqc.zip | (Illumina Only) A zip file containing the complete set of fastqc results |
| <SAMPLE_NAME>-{final|original}_fastp.json | (Illumina Only) A JSON file containing the QC results generated by fastp |
| <SAMPLE_NAME>-{final|original}_fastp.html | (Illumina Only) A HTML report of the QC results generated by fastp |
| <SAMPLE_NAME>-{final|original}_NanoPlot-report.html | (ONT Only) A HTML report of the QC results generated by NanoPlot |
| <SAMPLE_NAME>-{final|original}_NanoPlot.tar.gz | (ONT Only) A tarball containing the complete set of NanoPlot results |
Failed Quality Checks¶
Built into Bactopia are few basic quality checks to help prevent downstream failures. If a sample fails one of these checks, it will be excluded from further analysis. By excluding these samples, complete pipeline failures are prevented.
| Filename | Description |
|---|---|
| .error-fastq.gz | A gzipped FASTQ file of Illumina Single-End or Oxford Nanopore reads that failed QC |
| _R{1|2}.error-fastq.gz | A gzipped FASTQ file of Illumina Single-End or Oxford Nanopore reads that failed QC |
| -low-read-count-error.txt | Sample failed read count checks and excluded from further analysis |
| -low-sequence-coverage-error.txt | Sample failed sequenced coverage checks and excluded from further analysis |
| -low-sequence-depth-error.txt | Sample failed sequenced basepair checks and excluded from further analysis |
Poor samples are excluded to prevent downstream failures
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: After QC, too few reads remain
If after cleaning reads, a sample has less than the minimum required reads, the
sample will be excluded from further analysis. You can adjust this minimum read
count using the --min_reads parameter.
Example Text from <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Example Error: After QC, too little sequence coverage remains
If after cleaning reads, a sample has failed to meet the minimum sequence
coverage required, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_coverage parameter.
Note: This check is only performed when a genome size is available.
Example Text from <SAMPLE_NAME>-low-sequence-coverage-error.txt
After QC, <SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not
exceed the required minimum Y bp (Zx coverage). Further analysis is
discontinued.
Example Error: After QC, too little sequenced basepairs remain
If after cleaning reads, a sample has failed to meet the minimum number of
sequenced basepairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_basepairs parameter.
Example Text from <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
QC Parameters¶
| Parameter | Description |
|---|---|
--use_bbmap |
Illumina reads will be QC'd using BBMap Type: boolean |
--use_porechop |
Use Porechop to remove adapters from ONT reads Type: boolean, Default: turned off |
--skip_qc |
The QC step will be skipped and it will be assumed the inputs sequences have already been QCed. Type: boolean |
--skip_qc_plots |
QC Plot creation by FastQC or Nanoplot will be skipped Type: boolean |
--skip_error_correction |
FLASH error correction of reads will be skipped. Type: boolean |
--adapters |
A FASTA file containing adapters to remove Type: string |
--adapter_k |
Kmer length used for finding adapters. Type: integer, Default: 23 |
--phix |
phiX174 reference genome to remove Type: string |
--phix_k |
Kmer length used for finding phiX174. Type: integer, Default: 31 |
--ktrim |
Trim reads to remove bases matching reference kmers Type: string, Default: r |
--mink |
Look for shorter kmers at read tips down to this length, when k-trimming or masking. Type: integer, Default: 11 |
--hdist |
Maximum Hamming distance for ref kmers (subs only) Type: integer, Default: 1 |
--tpe |
When kmer right-trimming, trim both reads to the minimum length of either Type: string, Default: t |
--tbo |
Trim adapters based on where paired reads overlap Type: string, Default: t |
--qtrim |
Trim read ends to remove bases with quality below trimq. Type: string, Default: rl |
--trimq |
Regions with average quality BELOW this will be trimmed if qtrim is set to something other than f Type: integer, Default: 6 |
--maq |
Reads with average quality (after trimming) below this will be discarded Type: integer, Default: 10 |
--minlength |
Reads shorter than this after trimming will be discarded Type: integer, Default: 35 |
--ftm |
If positive, right-trim length to be equal to zero, modulo this number Type: integer, Default: 5 |
--tossjunk |
Discard reads with invalid characters as bases Type: string, Default: t |
--ain |
When detecting pair names, allow identical names Type: string, Default: f |
--qout |
PHRED offset to use for output FASTQs Type: string, Default: 33 |
--maxcor |
Max number of corrections within a 20bp window Type: integer, Default: 1 |
--sampleseed |
Set to a positive number to use as the random number generator seed for sampling Type: integer, Default: 42 |
--ont_minlength |
ONT Reads shorter than this will be discarded Type: integer, Default: 1000 |
--ont_minqual |
Minimum average read quality filter of ONT reads Type: integer |
--porechop_opts |
Extra Porechop options in quotes Type: integer |
--nanoplot_opts |
Extra NanoPlot options in quotes Type: integer |
--bbduk_opts |
Extra BBDuk options in quotes Type: integer |
--fastp_opts |
Extra fastp options in quotes Type: integer |
Citations¶
If you use Bactopia and qc results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
BBTools
Bushnell B BBMap short read aligner, and other bioinformatic tools. (Link) -
fastp
Chen S, Zhou Y, Chen Y, and Gu J fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884–i890. (2018) -
fastq-scan
Petit III RA fastq-scan: generate summary statistics of input FASTQ sequences. (GitHub) -
FastQC
Andrews S FastQC: a quality control tool for high throughput sequence data. (WebLink) -
Lighter
Song L, Florea L, Langmead B Lighter: Fast and Memory-efficient Sequencing Error Correction without Counting. Genome Biol. 15(11):509 (2014) -
NanoPlot
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C NanoPack: visualizing and processing long-read sequencing data Bioinformatics Volume 34, Issue 15 (2018) -
Nanoq
Steinig E Nanoq: Minimal but speedy quality control for nanopore reads in Rust (GitHub) -
Pigz
Adler M. pigz: A parallel implementation of gzip for modern multi-processor, multi-core machines. Jet Propulsion Laboratory (2015) -
Porechop
Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom. 3(10):e000132 (2017) -
Rasusa
Hall MB Rasusa: Randomly subsample sequencing reads to a specified coverage. (2019).
Step 3 - Assembler¶
The assembler module uses a variety of assembly tools to create an assembly of
Illumina and Oxford Nanopore reads. The tools used are:
| Tool | Description |
|---|---|
| Dragonflye | Assembly of Oxford Nanopore reads, as well as hybrid assembly with short-read polishing |
| Shovill | Assembly of Illumina paired-end reads |
| Shovill-SE | Assembly of Illumina single-end reads |
| Unicycler | Hybrid assembly, using short-reads first then long-reads |
Summary statistics for each assembly are generated using assembly-scan.
Outputs¶
Merged Results¶
Below are results that are concatenated into a single file.
| Filename | Description |
|---|---|
| assembly-scan.tsv | Assembly statistics for all samples |
Dragonflye¶
Below is a description of the per-sample results for Oxford Nanopore reads using Dragonflye.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.fna.gz | The final assembly produced by Dragonflye |
| <SAMPLE_NAME>.tsv | A tab-delimited file containing assembly statistics |
| flye-info.txt | A log file containing information about the Flye assembly |
| {flye|miniasm|raven}-unpolished.fasta.gz | Raw unprocessed assembly produced by the used assembler |
| {flye|miniasm|raven}-unpolished.gfa.gz | Raw unprocessed assembly graph produced by the used assembler |
Shovill¶
Below is a description of the per-sample results for Illumina reads using Shovill or Shovill-SE.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.fna.gz | The final assembly produced by Dragonflye |
| <SAMPLE_NAME>.tsv | A tab-delimited file containing assembly statistics |
| flash.hist | (Paired-End Only) Numeric histogram of merged read lengths. |
| flash.histogram | (Paired-End Only) Visual histogram of merged read lengths |
| {megahit|spades|velvet}-unpolished.gfa.gz | Raw unprocessed assembly graph produced by the used assembler |
| shovill.corrections | List of post-assembly corrections made by Shovill |
Hybrid Assembly (Unicycler)¶
Below is a description of the per-sample results for a hybrid assembly using
Unicycler (--hybrid). When using Unicycler,
the short-reads are assembled first, then the long-reads are used to polish the
assembly.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.fna.gz | The final assembly produced by Dragonflye |
| <SAMPLE_NAME>.tsv | A tab-delimited file containing assembly statistics |
| unicycler-unpolished.fasta.gz | Raw unprocessed assembly produced by Unicycler |
| unicycler-unpolished.fasta.gz | Raw unprocessed assembly graph produced by Unicycler |
Hybrid Assembly (Short Read Polishing)¶
Below is a description of the per-sample results for a hybrid assembly using
Dragonflye (--short_polish). When using
Dragonflye, the long-reads are assembled first, then the short-reads are used
to polish the assembly.
Prefer --short_polish over --hybrid with recent ONT sequencing
Using Unicycler (--hybrid) to create a hybrid
assembly works great when you have low-coverage noisy long-reads. However, if you are
using recent ONT sequencing, you likely have high-coverage and using the --short_polish
method is going to yeild better results (and be faster!) than --hybrid.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.fna.gz | The final assembly produced by Dragonflye |
| <SAMPLE_NAME>.tsv | A tab-delimited file containing assembly statistics |
| flye-info.txt | A log file containing information about the Flye assembly |
| {flye|miniasm|raven}-unpolished.fasta.gz | Raw unprocessed assembly produced by the used assembler |
| {flye|miniasm|raven}-unpolished.gfa.gz | Raw unprocessed assembly graph produced by the used assembler |
Failed Quality Checks¶
Built into Bactopia are few basic quality checks to help prevent downstream failures. If a sample fails one of these checks, it will be excluded from further analysis. By excluding these samples, complete pipeline failures are prevented.
| Filename | Description |
|---|---|
| -assembly-error.txt | Sample failed read count checks and excluded from further analysis |
Poor samples are excluded to prevent downstream failures
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: Assembled Successfully, but 0 Contigs
If a sample assembles successfully, but 0 contigs are formed, the sample will be excluded from further analysis.
Example Text from <SAMPLE_NAME>-assembly-error.txt
<SAMPLE_NAME> assembled successfully, but 0 contigs were formed. Please investigate
<SAMPLE_NAME> to determine a cause (e.g. metagenomic, contaminants, etc...) for this
outcome. Further assembly-based analysis of <SAMPLE_NAME> will be discontinued.
Example Error: Assembled successfully, but poor assembly size
If you sample assembles successfully, but the assembly size is less than the minimum
allowed genome size, the sample will be excluded from further analysis. You can
adjust this minimum size using the --min_genome_size parameter.
Example Text from <SAMPLE_NAME>-assembly-error.txt
<SAMPLE_NAME> assembled size (000 bp) is less than the minimum allowed genome
size (000 bp). If this is unexpected, please investigate <SAMPLE_NAME> to
determine a cause (e.g. metagenomic, contaminants, etc...) for the poor assembly.
Otherwise, adjust the --min_genome_size parameter to fit your need. Further
assembly based analysis of <SAMPLE_NAME> will be discontinued.
Assembler Parameters¶
| Parameter | Description |
|---|---|
--shovill_assembler |
Assembler to be used by Shovill Type: string, Default: skesa |
--dragonflye_assembler |
Assembler to be used by Dragonflye Type: string, Default: flye |
--use_unicycler |
Use unicycler for paired end assembly Type: boolean |
--min_contig_len |
Minimum contig length <0=AUTO> Type: integer, Default: 500 |
--min_contig_cov |
Minimum contig coverage <0=AUTO> Type: integer, Default: 2 |
--contig_namefmt |
Format of contig FASTA IDs in 'printf' style Type: string |
--shovill_opts |
Extra assembler options in quotes for Shovill Type: string |
--shovill_kmers |
K-mers to use Type: string |
--dragonflye_opts |
Extra assembler options in quotes for Dragonflye Type: string |
--trim |
Enable adaptor trimming Type: boolean |
--no_stitch |
Disable read stitching for paired-end reads Type: boolean |
--no_corr |
Disable post-assembly correction Type: boolean |
--unicycler_mode |
Bridging mode used by Unicycler Type: string, Default: normal |
--min_polish_size |
Contigs shorter than this value (bp) will not be polished using Pilon Type: integer, Default: 10000 |
--min_component_size |
Graph dead ends smaller than this size (bp) will be removed from the final graph Type: integer, Default: 1000 |
--min_dead_end_size |
Graph dead ends smaller than this size (bp) will be removed from the final graph Type: integer, Default: 1000 |
--nanohq |
For Flye, use '--nano-hq' instead of --nano-raw Type: boolean |
--medaka_model |
The model to use for Medaka polishing Type: string |
--medaka_rounds |
The number of Medaka polishing rounds to conduct Type: integer |
--racon_rounds |
The number of Racon polishing rounds to conduct Type: integer, Default: 1 |
--no_polish |
Skip the assembly polishing step Type: boolean |
--no_miniasm |
Skip miniasm+Racon bridging Type: boolean |
--no_rotate |
Do not rotate completed replicons to start at a standard gene Type: boolean |
--reassemble |
If reads were simulated, they will be used to create a new assembly. Type: boolean |
--polypolish_rounds |
Number of polishing rounds to conduct with Polypolish for short read polishing Type: integer, Default: 1 |
--pilon_rounds |
Number of polishing rounds to conduct with Pilon for short read polishing Type: integer |
Citations¶
If you use Bactopia and assembler results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
any2fasta
Seemann T any2fasta: Convert various sequence formats to FASTA (GitHub) -
assembly-scan
Petit III RA assembly-scan: generate basic stats for an assembly (GitHub) -
BWA
Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN] (2013) -
csvtk
Shen, W csvtk: A cross-platform, efficient and practical CSV/TSV toolkit in Golang. (GitHub) -
Dragonflye
Petit III RA Dragonflye: Assemble bacterial isolate genomes from Nanopore reads. (GitHub) -
FLASH
Magoč T, Salzberg SL FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27.21 2957-2963 (2011) -
Flye
Kolmogorov M, Yuan J, Lin Y, Pevzner P Assembly of Long Error-Prone Reads Using Repeat Graphs Nature Biotechnology (2019) -
Medaka
ONT Research Medaka: Sequence correction provided by ONT Research (GitHub) -
MEGAHIT
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31.10 1674-1676 (2015) -
Miniasm
Li H Miniasm: Ultrafast de novo assembly for long noisy reads (GitHub) -
Minimap2
Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094-3100 (2018) -
Nanoq
Steinig E Nanoq: Minimal but speedy quality control for nanopore reads in Rust (GitHub) -
Pigz
Adler M. pigz: A parallel implementation of gzip for modern multi-processor, multi-core machines. Jet Propulsion Laboratory (2015) -
Pilon
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9.11 e112963 (2014) -
Racon
Vaser R, Sović I, Nagarajan N, Šikić M Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017) -
Rasusa
Hall MB Rasusa: Randomly subsample sequencing reads to a specified coverage. (2019). -
Raven
Vaser R, Šikić M Time- and memory-efficient genome assembly with Raven. Nat Comput Sci 1, 332–336 (2021) -
samclip
Seemann T Samclip: Filter SAM file for soft and hard clipped alignments (GitHub) -
Samtools
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009) -
Shovill
Seemann T Shovill: De novo assembly pipeline for Illumina paired reads (GitHub) -
Shovill-SE
Petit III RA Shovill-SE: A fork of Shovill that includes support for single end reads. (GitHub) -
SKESA
Souvorov A, Agarwala R, Lipman DJ SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biology 19:153 (2018) -
SPAdes
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology 19.5 455-477 (2012) -
Unicycler
Wick RR, Judd LM, Gorrie CL, Holt KE Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017) -
Velvet
Zerbino DR, Birney E Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18.5 821-829 (2008)
Step 4 - Annotator¶
Outputs¶
Prokka¶
Below is a description of the per-sample results from Prokka.
| Filename | Description |
|---|---|
| .blastdb.tar.gz | A gzipped tar archive of BLAST+ database of the contigs, genes, and proteins |
| .faa.gz | Protein FASTA file of the translated CDS sequences. |
| .ffn.gz | Nucleotide FASTA file of all the prediction transcripts (CDS, rRNA, tRNA, tmRNA, misc_RNA) |
| .fna.gz | Nucleotide FASTA file of the input contig sequences. |
| .gbk.gz | This is a standard GenBank file derived from the master .gff. If the input to prokka was a multi-FASTA, then this will be a multi-GenBank, with one record for each sequence. |
| .gff.gz | This is the master annotation in GFF3 format, containing both sequences and annotations. It can be viewed directly in Artemis or IGV. |
| .sqn.gz | An ASN1 format "Sequin" file for submission to GenBank. It needs to be edited to set the correct taxonomy, authors, related publication etc. |
| .tbl.gz | Feature Table file, used by "tbl2asn" to create the .sqn file. |
| .tsv | Tab-separated file of all features (locus_tag,ftype,len_bp,gene,EC_number,COG,product) |
| .txt | Statistics relating to the annotated features found. |
Bakta¶
Below is a description of the per-sample results from Bakta.
| Filename | Description |
|---|---|
| .blastdb.tar.gz | A gzipped tar archive of BLAST+ database of the contigs, genes, and proteins |
| .embl.gz | Annotations & sequences in (multi) EMBL format |
| .faa.gz | CDS/sORF amino acid sequences as FASTA |
| .ffn.gz | Feature nucleotide sequences as FASTA |
| .fna.gz | Replicon/contig DNA sequences as FASTA |
| .gbff.gz | Annotations & sequences in (multi) GenBank format |
| .gff3.gz | Annotations & sequences in GFF3 format |
| .hypotheticals.faa.gz | Hypothetical protein CDS amino acid sequences as FASTA |
| .hypotheticals.tsv | Further information on hypothetical protein CDS as simple human readable tab separated values |
| .tsv | Annotations as simple human readable tab separated values |
| .txt | Broad summary of Bakta annotations |
Parameters¶
Prokka¶
| Parameter | Description |
|---|---|
--proteins |
FASTA file of trusted proteins to first annotate from Type: string |
--prodigal_tf |
Training file to use for Prodigal Type: string |
--compliant |
Force Genbank/ENA/DDJB compliance Type: boolean |
--centre |
Sequencing centre ID Type: string, Default: Bactopia |
--prokka_coverage |
Minimum coverage on query protein Type: integer, Default: 80 |
--prokka_evalue |
Similarity e-value cut-off Type: string, Default: 1e-09 |
--prokka_opts |
Extra Prokka options in quotes. Type: string |
Bakta Download¶
| Parameter | Description |
|---|---|
--bakta_db |
Tarball or path to the Bakta database Type: string |
--bakta_db_type |
Which Bakta DB to download 'full' (~30GB) or 'light' (~2GB) Type: string, Default: full |
--bakta_save_as_tarball |
Save the Bakta database as a tarball Type: boolean |
--download_bakta |
Download the Bakta database to the path given by --bakta_db Type: boolean |
Bakta¶
| Parameter | Description |
|---|---|
--proteins |
FASTA file of trusted proteins to first annotate from Type: string |
--prodigal_tf |
Training file to use for Prodigal Type: string |
--replicons |
Replicon information table (tsv/csv) Type: string |
--min_contig_length |
Minimum contig size to annotate Type: integer, Default: 1 |
--keep_contig_headers |
Keep original contig headers Type: boolean |
--compliant |
Force Genbank/ENA/DDJB compliance Type: boolean |
--skip_trna |
Skip tRNA detection & annotation Type: boolean |
--skip_tmrna |
Skip tmRNA detection & annotation Type: boolean |
--skip_rrna |
Skip rRNA detection & annotation Type: boolean |
--skip_ncrna |
Skip ncRNA detection & annotation Type: boolean |
--skip_ncrna_region |
Skip ncRNA region detection & annotation Type: boolean |
--skip_crispr |
Skip CRISPR array detection & annotation Type: boolean |
--skip_cds |
Skip CDS detection & annotation Type: boolean |
--skip_sorf |
Skip sORF detection & annotation Type: boolean |
--skip_gap |
Skip gap detection & annotation Type: boolean |
--skip_ori |
Skip oriC/oriT detection & annotation Type: boolean |
--bakta_opts |
Extra Backa options in quotes. Example: '--gram +' Type: string |
Citations¶
If you use Bactopia and annotator results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
Prokka
Seemann T Prokka: rapid prokaryotic genome annotation Bioinformatics 30, 2068–2069 (2014) -
Bakta
Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A Bakta - rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microbial Genomics 7(11) (2021)
Step 5 - Sketcher¶
The sketcher module uses Mash and
Sourmash to create sketches and query
RefSeq and GTDB.
Outputs¶
sketcher¶
Below is a description of the per-sample results from the sketcher subworkflow.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>-k{21|31}.msh | A Mash sketch of the input assembly for k=21 and k=31 |
| <SAMPLE_NAME>-mash-refseq88-k21.txt | The results of querying the Mash sketch against RefSeq88 |
| <SAMPLE_NAME>-sourmash-gtdb-rs207-k31.txt | The results of querying the Sourmash sketch against GTDB-rs207 |
| <SAMPLE_NAME>.sig | A Sourmash sketch of the input assembly for k=21, k=31, and k=51 |
Sketcher Parameters¶
| Parameter | Description |
|---|---|
--sketch_size |
Sketch size. Each sketch will have at most this many non-redundant min-hashes. Type: integer, Default: 10000 |
--sourmash_scale |
Choose number of hashes as 1 in FRACTION of input k-mers Type: integer, Default: 10000 |
--no_winner_take_all |
Disable winner-takes-all strategy for identity estimates Type: boolean |
--screen_i |
Minimum identity to report. Type: number, Default: 0.8 |
Citations¶
If you use Bactopia and sketcher results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
Genome Taxonomy Database
Parks DH, Chuvochina M, Rinke C, Mussig AJ, Chaumeil P-A, Hugenholtz P GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy Nucleic Acids Research gkab776 (2021) -
Mash
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17, 132 (2016) -
Mash
Ondov BD, Starrett GJ, Sappington A, Kostic A, Koren S, Buck CB, Phillippy AM Mash Screen: high-throughput sequence containment estimation for genome discovery Genome Biol 20, 232 (2019) -
NCBI RefSeq Database
O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O0, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O'Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45 (2016) -
Sourmash
Brown CT, Irber L sourmash: a library for MinHash sketching of DNA. JOSS 1, 27 (2016)
Step 6 - Sequence Typing¶
Outputs¶
Merged Results¶
Below are results that are concatenated into a single file.
| Filename | Description |
|---|---|
| mlst.tsv | A merged TSV file with mlst results from all samples |
mlst¶
Below is a description of the per-sample results from mlst.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with mlst result, see mlst - Usage for more details |
Parameters¶
| Parameter | Description |
|---|---|
--scheme |
Don't autodetect, force this scheme on all inputs Type: string |
--minid |
Minimum DNA percent identity of full allelle to consider 'similar' Type: integer, Default: 95 |
--mincov |
Minimum DNA percent coverage to report partial allele at all Type: integer, Default: 10 |
--minscore |
Minumum score out of 100 to match a scheme Type: integer, Default: 50 |
--nopath |
Strip filename paths from FILE column Type: boolean |
Citations¶
If you use Bactopia and sequence typing results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
csvtk
Shen, W csvtk: A cross-platform, efficient and practical CSV/TSV toolkit in Golang. (GitHub) -
mlst
Seemann T mlst: scan contig files against PubMLST typing schemes (GitHub) -
PubMLST.org
Jolley KA, Bray JE, Maiden MCJ Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3, 124 (2018)
Step 7 - Antibiotic Resistance¶
Outputs¶
Merged Results¶
Below are results that are concatenated into a single file.
| Filename | Description |
|---|---|
| amrfinderplus-genes.tsv | A merged TSV file with AMRFinder+ results using nucleotide inputs |
| amrfinderplus-proteins.tsv | A merged TSV file with AMRFinder+ results using protein inputs |
AMRFinder+¶
Below is a description of the per-sample results from AMRFinder+.
| Filename | Description |
|---|---|
| -genes.tsv | A TSV file with AMRFinder+ results using nucleotide inputs |
| -proteins.tsv | A TSV file with AMRFinder+ results using protein inputs |
Parameters¶
| Parameter | Description |
|---|---|
--ident_min |
Minimum proportion of identical amino acids in alignment for hit (0..1) Type: number, Default: -1 |
--coverage_min |
Minimum coverage of the reference protein (0..1) Type: number, Default: 0.5 |
--organism |
Taxonomy group to run additional screens against Type: string |
--translation_table |
NCBI genetic code for translated BLAST Type: integer, Default: 11 |
--amrfinder_noplus |
Disable running AMRFinder+ with the --plus option Type: boolean |
--report_common |
Report proteins common to a taxonomy group Type: boolean |
--report_all_equal |
Report all equally-scoring BLAST and HMM matches Type: boolean |
--amrfinder_opts |
Extra AMRFinder+ options in quotes. Type: string |
Citations¶
If you use Bactopia and antibiotic resistance results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
AMRFinderPlus
Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, Tyson GH, Zhao S, Hsu C-H, McDermott PF, Tadesse DA, Morales C, Simmons M, Tillman G, Wasilenko J, Folster JP, Klimke W Validating the NCBI AMRFinder Tool and Resistance Gene Database Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of NARMS Isolates. Antimicrob. Agents Chemother. (2019) -
csvtk
Shen, W csvtk: A cross-platform, efficient and practical CSV/TSV toolkit in Golang. (GitHub)
Step 8 - Merlin¶
Outputs¶
Merged Results¶
Below are results that are concatenated into a single file.
| Filename | Description |
|---|---|
| agrvate.tsv | A merged TSV file with AgrVATE results from all samples |
| ectyper.tsv | A merged TSV file with ECTyper results from all samples |
| emmtyper.tsv | A merged TSV file with emmtyper results from all samples |
| genotyphi.tsv | A merged TSV file with genotyphi results from all samples |
| hicap.tsv | A merged TSV file with hicap results from all samples |
| hpsuissero.tsv | A merged TSV file with HpsuisSero results from all samples |
| kleborate.tsv | A merged TSV file with Kleborate results from all samples |
| legsta.tsv | A merged TSV file with legsta results from all samples |
| lissero.tsv | A merged TSV file with LisSero results from all samples |
| meningotype.tsv | A merged TSV file with meningotype results from all samples |
| ngmaster.tsv | A merged TSV file with ngmaster results from all samples |
| pasty.tsv | A merged TSV file with pasty results from all samples |
| pbptyper.tsv | A merged TSV file with pbptyper results from all samples |
| seqsero2.tsv | A merged TSV file with seqsero2 results from all samples |
| seroba.tsv | A merged TSV file with seroba results from all samples |
| shigatyper.tsv | A merged TSV file with ShigaTyper results from all samples |
| shigeifinder.tsv | A merged TSV file with ShigEiFinder results from all samples |
| sistr.tsv | A merged TSV file with SISTR results from all samples |
| spatyper.tsv | A merged TSV file with spaTyper results from all samples |
| ssuissero.tsv | A merged TSV file with SsuisSero results from all samples |
| staphopiasccmec.tsv | A merged TSV file with staphopia-sccmec results from all samples |
| stecfinder.tsv | A merged TSV file with stecfinder results from all samples |
AgrVATE¶
Below is a description of the per-sample results from AgrVATE.
| Filename | Description |
|---|---|
| -agr_gp.tab | A detailed report for agr kmer matches |
| -blastn_log.txt | Log files from programs called by AgrVATE |
| -summary.tab | A final summary report for agr typing |
ECTyper¶
Below is a description of the per-sample results from ECTyper.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with ECTyper result, see ECTyper - Report format for details |
| blast_output_alleles.txt | Allele report generated from BLAST results |
emmtyper¶
Below is a description of the per-sample results from emmtyper.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with emmtyper result, see emmtyper - Result format for details |
hicap¶
Below is a description of the per-sample results from hicap.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.gbk | GenBank file and cap locus annotations |
| <SAMPLE_NAME>.svg | Visualization of annotated cap locus |
| <SAMPLE_NAME>.tsv | A tab-delimited file with hicap results |
HpsuisSero¶
Below is a description of the per-sample results from HpsuisSero.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>_serotyping_res.tsv | A tab-delimited file with HpsuisSero result |
GenoTyphi¶
Below is a description of the per-sample results from GenoTyphi. A full description of the GenoTyphi output is available at GenoTyphi - Output
| Filename | Description |
|---|---|
| <SAMPLE_NAME>_predictResults.tsv | A tab-delimited file with GenoTyphi results |
| <SAMPLE_NAME>.csv | The output of mykrobe predict in comma-separated format |
| <SAMPLE_NAME>.json | The output of mykrobe predict in JSON format |
Kleborate¶
Below is a description of the per-sample results from Kleborate.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.results.txt | A tab-delimited file with Kleborate result, see Kleborate - Example output for more details. |
legsta¶
Below is a description of the per-sample results from legsta.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with legsta result, see legsta - Output for more details |
LisSero¶
Below is a description of the per-sample results from LisSero.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with LisSero results |
Mash¶
Below is a description of the per-sample results from Mash.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>-dist.txt | A tab-delimited file with mash dist results |
meningotype¶
Below is a description of the per-sample results from meningotype .
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with meningotype result |
ngmaster¶
Below is a description of the per-sample results from ngmaster.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with ngmaster results |
pasty¶
Below is a description of the per-sample results from pasty.
| Filename | Description |
|---|---|
| .blastn.tsv | A tab-delimited file of all blast hits |
| .details.tsv | A tab-delimited file with details for each serogroup |
| .tsv | A tab-delimited file with the predicted serogroup |
pbptyper¶
Below is a description of the per-sample results from pbptyper.
| Filename | Description |
|---|---|
| .tblastn.tsv | A tab-delimited file of all blast hits |
| .tsv | A tab-delimited file with the predicted PBP type |
SeqSero2¶
Below is a description of the per-sample results from SeqSero2.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>_result.tsv | A tab-delimited file with SeqSero2 results |
| <SAMPLE_NAME>_result.txt | A text file with key-value pairs of SeqSero2 results |
Seroba¶
Below is a description of the per-sample results from Seroba. More details about the outputs are available from Seroba - Output.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with the predicted serotype |
| detailed_serogroup_info.txt | Detailed information about the predicted results |
ShigaTyper¶
Below is a description of the per-sample results from ShigaTyyper.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>-hits.tsv | Detailed statistics about each individual gene hit |
| <SAMPLE_NAME>.tsv | The final predicted serotype by ShigaTyper |
ShigEiFinder¶
Below is a description of the per-sample results from ShigEiFinder.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with the predicted Shigella or EIEC serotype |
SISTR¶
Below is a description of the per-sample results from SISTR.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>-allele.fasta.gz | A FASTA file of the cgMLST allele search results |
| <SAMPLE_NAME>-allele.json.gz | JSON formated cgMLST allele search results, see SISTR - cgMLST search results for more details |
| <SAMPLE_NAME>-cgmlst.csv | A comma-delimited summary of the cgMLST allele search results |
| <SAMPLE_NAME>.tsv | A tab-delimited file with SISTR results, see SISTR - Primary results for more details |
spaTyper¶
Below is a description of the per-sample results from spaTyper.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with spaTyper result |
SsuisSero¶
Below is a description of the per-sample results from SsuisSero.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>_serotyping_res.tsv | A tab-delimited file with SsuisSero results |
staphopia-sccmec¶
Below is a description of the per-sample results from staphopia-sccmec.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.tsv | A tab-delimited file with staphopia-sccmec results |
TBProfiler¶
Below is a description of the per-sample results from TBProfiler.
| Filename | Description |
|---|---|
| <SAMPLE_NAME>.results.csv | A CSV formated TBProfiler result file of resistance and strain type |
| <SAMPLE_NAME>.results.json | A JSON formated TBProfiler result file of resistance and strain type |
| <SAMPLE_NAME>.results.txt | A text file with TBProfiler results |
| <SAMPLE_NAME>.bam | BAM file with alignment details |
| <SAMPLE_NAME>.targets.csq.vcf.gz | VCF with variant info again reference genomes |
¶
mashdist¶
| Parameter | Description |
|---|---|
--mash_sketch |
The reference sequence as a Mash Sketch (.msh file) Type: string |
--mash_seed |
Seed to provide to the hash function Type: integer, Default: 42 |
--mash_table |
Table output (fields will be blank if they do not meet the p-value threshold) Type: boolean |
--mash_m |
Minimum copies of each k-mer required to pass noise filter for reads Type: integer, Default: 1 |
--mash_w |
Probability threshold for warning about low k-mer size. Type: number, Default: 0.01 |
--max_p |
Maximum p-value to report. Type: number, Default: 1.0 |
--max_dist |
Maximum distance to report. Type: number, Default: 1.0 |
--merlin_dist |
Maximum distance to report when using Merlin . Type: number, Default: 0.1 |
--full_merlin |
Go full Merlin and run all species-specific tools, no matter the Mash distance Type: boolean |
--use_fastqs |
Query with FASTQs instead of the assemblies Type: boolean |
AgrVATE¶
| Parameter | Description |
|---|---|
--typing_only |
agr typing only. Skips agr operon extraction and frameshift detection Type: boolean |
ECTyper¶
| Parameter | Description |
|---|---|
--opid |
Percent identity required for an O antigen allele match Type: integer, Default: 90 |
--opcov |
Minumum percent coverage required for an O antigen allele match Type: integer, Default: 90 |
--hpid |
Percent identity required for an H antigen allele match Type: integer, Default: 95 |
--hpcov |
Minumum percent coverage required for an H antigen allele match Type: integer, Default: 50 |
--verify |
Enable E. coli species verification Type: boolean |
--print_alleles |
Prints the allele sequences if enabled as the final column Type: boolean |
emmtyper¶
| Parameter | Description |
|---|---|
--emmtyper_wf |
Workflow for emmtyper to use. Type: string, Default: blast |
--cluster_distance |
Distance between cluster of matches to consider as different clusters Type: integer, Default: 500 |
--percid |
Minimal percent identity of sequence Type: integer, Default: 95 |
--culling_limit |
Total hits to return in a position Type: integer, Default: 5 |
--mismatch |
Threshold for number of mismatch to allow in BLAST hit Type: integer, Default: 5 |
--align_diff |
Threshold for difference between alignment length and subject length in BLAST Type: integer, Default: 5 |
--gap |
Threshold gap to allow in BLAST hit Type: integer, Default: 2 |
--min_perfect |
Minimum size of perfect match at 3 primer end Type: integer, Default: 15 |
--min_good |
Minimum size where there must be 2 matches for each mismatch Type: integer, Default: 15 |
--max_size |
Maximum size of PCR product Type: integer, Default: 2000 |
hicap¶
| Parameter | Description |
|---|---|
--database_dir |
Directory containing locus database Type: string |
--model_fp |
Path to prodigal model Type: string |
--full_sequence |
Write the full input sequence out to the genbank file rather than just the region surrounding and including the locus Type: boolean |
--hicap_debug |
hicap will print debug messages Type: boolean |
--gene_coverage |
Minimum percentage coverage to consider a single gene complete Type: number, Default: 0.8 |
--gene_identity |
Minimum percentage identity to consider a single gene complete Type: number, Default: 0.7 |
--broken_gene_length |
Minimum length to consider a broken gene Type: integer, Default: 60 |
--broken_gene_identity |
Minimum percentage identity to consider a broken gene Type: number, Default: 0.8 |
GenoTyphi¶
| Parameter | Description |
|---|---|
--kmer |
K-mer length Type: integer, Default: 21 |
--min_depth |
Minimum depth Type: integer, Default: 1 |
--model |
Genotype model used. Type: string, Default: kmer_count |
--report_all_calls |
Report all calls Type: boolean |
--mykrobe_opts |
Extra Mykrobe options in quotes Type: string |
Kleborate¶
| Parameter | Description |
|---|---|
--skip_resistance |
Turn off resistance genes screening Type: boolean |
--skip_kaptive |
Turn off Kaptive screening of K and O loci Type: boolean |
--min_identity |
Minimum alignment percent identity for main results Type: number, Default: 90.0 |
--kleborate_min_coverage |
Minimum alignment percent coverage for main results Type: number, Default: 80.0 |
--min_spurious_identity |
Minimum alignment percent identity for spurious results Type: number, Default: 80.0 |
--min_spurious_coverage |
Minimum alignment percent coverage for spurious results Type: number, Default: 40.0 |
--min_kaptive_confidence |
Minimum Kaptive confidence to call K/O loci - confidence levels below this will be reported as unknown Type: string, Default: Good |
--force_index |
Rebuild the BLAST index at the start of execution Type: boolean |
legsta¶
| Parameter | Description |
|---|---|
--noheader |
Don't print header row Type: boolean |
LisSero¶
| Parameter | Description |
|---|---|
--min_id |
Minimum percent identity to accept a match Type: number, Default: 95.0 |
--min_cov |
Minimum coverage of the gene to accept a match Type: number, Default: 95.0 |
meningotype¶
You can use these parameters to fine-tune your meningotype analysis
| Parameter | Description |
|---|---|
--finetype |
perform porA and fetA fine typing Type: boolean |
--porB |
perform porB sequence typing (NEIS2020) Type: boolean |
--bast |
perform Bexsero antigen sequence typing (BAST) Type: boolean |
--mlst |
perform MLST Type: boolean |
--all |
perform MLST, porA, fetA, porB, BAST typing Type: boolean |
ngmaster¶
| Parameter | Description |
|---|---|
--csv |
output comma-separated format (CSV) rather than tab-separated Type: boolean |
pasty¶
| Parameter | Description |
|---|---|
--pasty_min_pident |
Minimum percent identity to count a hit Type: integer, Default: 95 |
--pasty_min_coverage |
Minimum percent coverage to count a hit Type: integer, Default: 95 |
pbptyper¶
| Parameter | Description |
|---|---|
--pbptyper_min_pident |
Minimum percent identity to count a hit Type: integer, Default: 95 |
--pbptyper_min_coverage |
Minimum percent coverage to count a hit Type: integer, Default: 95 |
--pbptyper_min_ani |
Minimum S. pneumoniae ANI to predict PBP Type Type: integer, Default: 95 |
SeqSero2¶
| Parameter | Description |
|---|---|
--run_mode |
Workflow to run. 'a' allele mode, or 'k' k-mer mode Type: string, Default: k |
--input_type |
Input format to analyze. 'assembly' or 'fastq' Type: string, Default: assembly |
--bwa_mode |
Algorithms for bwa mapping for allele mode Type: string, Default: mem |
SISTR¶
| Parameter | Description |
|---|---|
--full_cgmlst |
Use the full set of cgMLST alleles which can include highly similar alleles Type: boolean |
spaTyper¶
| Parameter | Description |
|---|---|
--repeats |
List of spa repeats Type: string |
--repeat_order |
List spa types and order of repeats Type: string |
--do_enrich |
Do PCR product enrichment Type: boolean |
staphopia-sccmec¶
| Parameter | Description |
|---|---|
--hamming |
Report the results as hamming distances Type: boolean |
TBProfiler¶
| Parameter | Description |
|---|---|
--call_whole_genome |
Call whole genome Type: boolean |
--mapper |
Mapping tool to use. If you are using nanopore data it will default to minimap2 Type: string, Default: bwa |
--caller |
Variant calling tool to use Type: string, Default: freebayes |
--calling_params |
Extra variant caller options in quotes Type: string |
--suspect |
Use the suspect suite of tools to add ML predictions Type: boolean |
--no_flagstat |
Don't collect flagstats Type: boolean |
--no_delly |
Don't run delly Type: boolean |
--tbprofiler_opts |
Extra options in quotes for TBProfiler Type: string |
Citations¶
If you use Bactopia and merlin results in your analysis, please cite the following.
-
Bactopia
Petit III RA, Read TD Bactopia - a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (2020) -
AgrVATE
Raghuram V. AgrVATE: Rapid identification of Staphylococcus aureus agr locus type and agr operon variants. (GitHub) -
csvtk
Shen, W csvtk: A cross-platform, efficient and practical CSV/TSV toolkit in Golang. (GitHub) -
ECTyper
Laing C, Bessonov K, Sung S, La Rose C ECTyper - In silico prediction of Escherichia coli serotype (GitHub) -
emmtyper
Tan A, Seemann T, Lacey D, Davies M, Mcintyre L, Frost H, Williamson D, Gonçalves da Silva A emmtyper - emm Automatic Isolate Labeller (GitHub) -
GenoTyphi
Wong VK, Baker S, Connor TR, Pickard D, Page AJ, Dave J, Murphy N, Holliman R, Sefton A, Millar M, Dyson ZA, Dougan G, Holt KE, & International Typhoid Consortium. An extended genotyping framework for Salmonella enterica serovar Typhi, the cause of human typhoid Nature Communications 7, 12827. (2016) -
hicap
Watts SC, Holt KE hicap: in silico serotyping of the Haemophilus influenzae capsule locus. Journal of Clinical Microbiology JCM.00190-19 (2019) -
HpsuisSero
Lui J HpsuisSero: Rapid Haemophilus parasuis serotyping (GitHub) -
Kleborate
Lam MMC, Wick RR, Watts, SC, Cerdeira LT, Wyres KL, Holt KE A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat Commun 12, 4188 (2021) -
legsta
Seemann T legsta: In silico Legionella pneumophila Sequence Based Typing (GitHub) -
LisSero
Kwong J, Zhang J, Seeman T, Horan, K, Gonçalves da Silva A LisSero - In silico serotype prediction for Listeria monocytogenes (GitHub) -
Mash
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17, 132 (2016) -
meningotype
Kwong JC, Gonçalves da Silva A, Stinear TP, Howden BP, & Seemann T meningotype: in silico typing for Neisseria meningitidis. (GitHub) -
Mykrobe
Hunt M, Bradley P, Lapierre SG, Heys S, Thomsit M, Hall MB, Malone KM, Wintringer P, Walker TM, Cirillo DM, Comas I, Farhat MR, Fowler P, Gardy J, Ismail N, Kohl TA, Mathys V, Merker M, Niemann S, Omar SV, Sintchenko V, Smith G, Supply P, Tahseen S, Wilcox M, Arandjelovic I, Peto TEA, Crook, DW, Iqbal Z Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe Wellcome Open Research 4, 191. (2019) -
ngmaster
Kwong J, Gonçalves da Silva A, Schultz M, Seeman T ngmaster - In silico multi-antigen sequence typing for Neisseria gonorrhoeae (NG-MAST) (GitHub) -
pasty
Petit III RA pasty: in silico serogrouping of Pseudomonas aeruginosa isolates (GitHub) -
pbptyper
Petit III RA pbptyper: In silico Penicillin Binding Protein (PBP) typer for Streptococcus pneumoniae assemblies (GitHub) -
SeqSero2
Zhang S, Den-Bakker HC, Li S, Dinsmore BA, Lane C, Lauer AC, Fields PI, Deng X. SeqSero2: rapid and improved Salmonella serotype determination using whole genome sequencing data. Appl Environ Microbiology 85(23):e01746-19 (2019) -
ShigaTyper
Wu Y, Lau HK, Lee T, Lau DK, Payne J In Silico Serotyping Based on Whole-Genome Sequencing Improves the Accuracy of Shigella Identification. Applied and Environmental Microbiology, 85(7). (2019) -
ShigEiFinder
Zhang X, Payne M, Nguyen T, Kaur S, Lan R Cluster-specific gene markers enhance Shigella and enteroinvasive Escherichia coli in silico serotyping. Microbial Genomics, 7(12). (2021) -
SISTR
Yoshida CE, Kruczkiewicz P, Laing CR, Lingohr EJ, Gannon VPJ, Nash JHE, Taboada EN The Salmonella In Silico Typing Resource (SISTR): An Open Web-Accessible Tool for Rapidly Typing and Subtyping Draft Salmonella Genome Assemblies. PloS One, 11(1), e0147101. (2016) -
spaTyper
Sanchez-Herrero JF, and Sullivan M spaTyper: Staphylococcal protein A (spa) characterization pipeline. Zenodo. (2020) -
SsuisSero
Lui J SsuisSero: Rapid Streptococcus suis serotyping (GitHub) -
staphopia-sccmec
Petit III RA, Read TD Staphylococcus aureus viewed from the perspective of 40,000+ genomes. PeerJ 6, e5261 (2018) -
TBProfiler
Phelan JE, O’Sullivan DM, Machado D, Ramos J, Oppong YEA, Campino S, O’Grady J, McNerney R, Hibberd ML, Viveiros M, Huggett JF, Clark TG Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med 11, 41 (2019)
Additional Parameters¶
Optional¶
These optional parameters can be useful in certain settings.
| Parameter | Description |
|---|---|
--outdir |
Base directory to write results to Type: string, Default: ./ |
--run_name |
Name of the directory to hold results Type: string, Default: bactopia |
--skip_compression |
Ouput files will not be compressed Type: boolean |
--datasets |
The path to cache datasets to Type: string |
--keep_all_files |
Keeps all analysis files created Type: boolean |
Max Job Request¶
Set the top limit for requested resources for any single job.
| Parameter | Description |
|---|---|
--max_retry |
Maximum times to retry a process before allowing it to fail. Type: integer, Default: 3 |
--max_cpus |
Maximum number of CPUs that can be requested for any single job. Type: integer, Default: 4 |
--max_memory |
Maximum amount of memory (in GB) that can be requested for any single job. Type: integer, Default: 32 |
--max_time |
Maximum amount of time (in minutes) that can be requested for any single job. Type: integer, Default: 120 |
--max_downloads |
Maximum number of samples to download at a time Type: integer, Default: 3 |
Nextflow Configuration¶
to fine-tune your Nextflow setup.
| Parameter | Description |
|---|---|
--nfconfig |
A Nextflow compatible config file for custom profiles, loaded last and will overwrite existing variables if set. Type: string |
--publish_dir_mode |
Method used to save pipeline results to output directory. Type: string, Default: copy |
--infodir |
Directory to keep pipeline Nextflow logs and reports. Type: string, Default: ${params.outdir}/pipeline_info |
--force |
Nextflow will overwrite existing output files. Type: boolean |
--cleanup_workdir |
After Bactopia is successfully executed, the work directory will be deleted. Type: boolean |
Nextflow Profile¶
to fine-tune your Nextflow setup.
| Parameter | Description |
|---|---|
--condadir |
Directory to Nextflow should use for Conda environments Type: string |
--registry |
Docker registry to pull containers from. Type: string, Default: dockerhub |
--datasets_cache |
Directory where downloaded datasets should be stored. Type: string, Default: <BACTOPIA_DIR>/data/datasets |
--singularity_cache |
Directory where remote Singularity images are stored. Type: string |
--singularity_pull_docker_container |
Instead of directly downloading Singularity images for use with Singularity, force the workflow to pull and convert Docker containers instead. Type: boolean |
--force_rebuild |
Force overwrite of existing pre-built environments. Type: boolean |
--queue |
Comma-separated name of the queue(s) to be used by a job scheduler (e.g. AWS Batch or SLURM) Type: string, Default: general,high-memory |
--cluster_opts |
Additional options to pass to the executor. (e.g. SLURM: '--account=my_acct_name' Type: string |
--disable_scratch |
All intermediate files created on worker nodes of will be transferred to the head node. Type: boolean |
Helpful¶
Uncommonly used parameters that might be useful.
| Parameter | Description |
|---|---|
--monochrome_logs |
Do not use coloured log outputs. Type: boolean |
--nfdir |
Print directory Nextflow has pulled Bactopia to Type: boolean |
--sleep_time |
The amount of time (seconds) Nextflow will wait after setting up datasets before execution. Type: integer, Default: 5 |
--validate_params |
Boolean whether to validate parameters against the schema at runtime Type: boolean, Default: True |
--help |
Display help text. Type: boolean |
--wf |
Specify which workflow or Bactopia Tool to execute Type: string, Default: bactopia |
--list_wfs |
List the available workflows and Bactopia Tools to use with '--wf' Type: boolean |
--show_hidden_params |
Show all params when using --help Type: boolean |
--help_all |
An alias for --help --show_hidden_params Type: boolean |
--version |
Display version text. Type: boolean |