Beginner's Guide
Bactopia is a complete pipeline for the analysis of bacterial genomes, which includes more than 150 bioinformatics tools. In this section, we will discuss the most essential parameters users will need to make use of to get started with Bactopia. We are going to focus on the parameters associated with processing input samples.
Looking at the workflow overview below, we are really going to focus on the first step, the Gather step. This step gets all the data in one place, whether its local FASTQs, FASTQs from SRA or ENA, or assemblies from NCBI Assembly. The following guide will provide few examples of each of these accepted inputs, including:
- Local Illumina and/or Nanopore Reads
- Local Assemblies
- ENA/SRA Experiment Accessions
- NCBI Assembly Accessions
Towards the end of this guide, we'll also take a look at some helpful parameters. If you are interested in learning more about the full set of parameters available in Bactopia, please check out the Full Guide section.
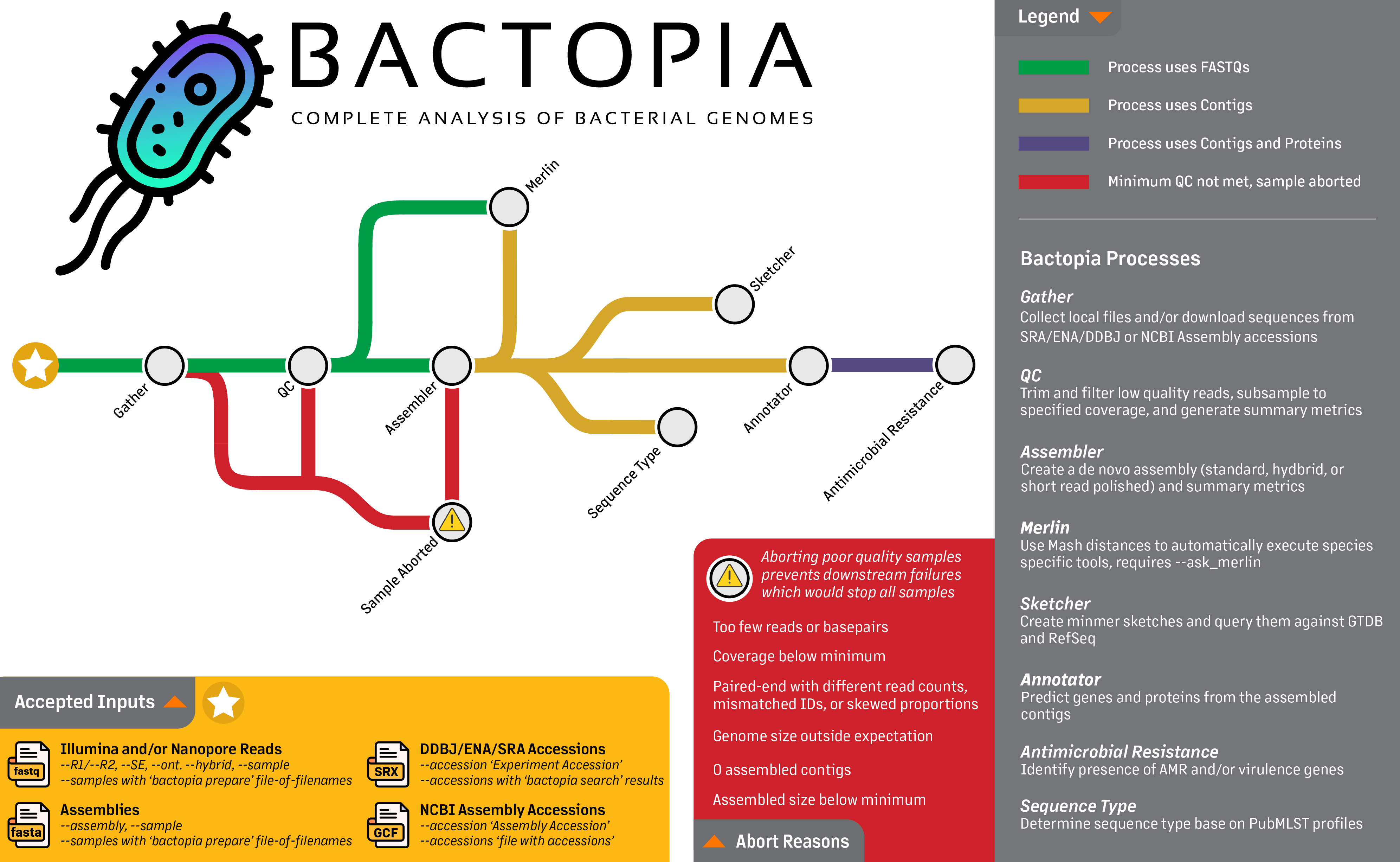
Gathering Inputs¶
Below is a table of essential parameters you will need to get started using Bactopia. This does not mean you need to use them all at once, but it will be useful to familiarize yourself with them. We will start here, with a brief description of each parameter, then we will go into more detail about each with example use cases.
Input Parameters¶
The following parameters are how you will provide either local or remote samples to be processed by Bactopia.
| Parameter | Description |
|---|---|
--samples |
A FOFN (via bactopia prepare) with sample names and paths to FASTQ/FASTAs to process Type: string |
--r1 |
First set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r2 and --sample) Type: string |
--r2 |
Second set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r1 and --sample) Type: string |
--se |
Compressed (gzip) Illumina single-end FASTQ reads (requires --sample) Type: string |
--ont |
Compressed (gzip) Oxford Nanopore FASTQ reads (requires --sample) Type: string |
--hybrid |
Create hybrid assembly using Unicycler. (requires --r1, --r2, --ont and --sample) Type: boolean |
--short_polish |
Create hybrid assembly from long-read assembly and short read polishing. (requires --r1, --r2, --ont and --sample) Type: boolean |
--sample |
Sample name to use for the input sequences Type: string |
--accessions |
A file containing ENA/SRA Experiment accessions or NCBI Assembly accessions to processed Type: string |
--accession |
Sample name to use for the input sequences Type: string |
--assembly |
A assembled genome in compressed FASTA format. (requires --sample) Type: string |
--check_samples |
Validate the input FOFN provided by --samples Type: boolean |
Now let's take a look at each parameter in more detail with a few example use-cases.
Single Sample¶
It's no secret that Bactopia accepts many different types of inputs from a single entry point (e.g. you don't need a separate pipeline for each input type). For now we are going to look at local inputs. In other words, inputs that are already on the machine you will be running Bactopia on. We will look at the following inputs:
- Local Illumina and/or Nanopore Reads
- Local Assemblies
- Processing Multiple Samples
Illumina and/or Nanopore Reads¶
Let's start with the most common inputs, plain on FASTQ files for a single sample. Bactopia accepts both Illumina (pair-end or single-end) and Nanopore reads, and can even process them together for a hybrid assembly.
Again, here we are focussing on processing a single sample at a time. To do this, you have to
provide a combination of the sample name (--sample) and the input type:
| Input Type | Required Parameters |
|---|---|
| Illumina Paired-End | --r1 and --r2 |
| Illumina Single-End | --se |
| Oxford Nanopore | --ont |
| Hybrid | --r1, --r2, --ont, and --hybrid |
| Hybrid (Short-read Polishing) | --r1, --r2, --ont, and --short_polish |
--sample is always required for single-sample processing
When processing a single sample, you will always have to provide --sample, no matter
input type. This parameter is used to name the output files and directories.
Paired-End¶
In this example, Bactopia will process the sample as paired-end Illumina reads. The
--r1 and --r2 parameters are used to specify the location of the first and second
pair of reads. In addition, the value of --sample will be used as the prefix
(e.g. my-sample.fna.gz) for saving results.
Use --r1, --r2 for Paired-End Illumina Reads
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz
Single-End¶
In this example, Bactopia will process the sample as single-end Illumina reads. The
--se parameter is used to specify the location of the single-end reads. Again, the
value of --sample will be used as the prefix for saving results.
Use --se for Single-End Illumina Reads
bactopia \
--sample my-sample \
--se /path/to/my-sample.fastq.gz
Nanopore¶
Let's change pace a little, to process Nanopore reads you will need --ont to specify
the location of the Nanopore reads as well as --sample for naming outputs.
Use --ont for Oxford Nanopore Reads
bactopia \
--sample my-sample \
--ont /path/to/my-sample.fastq.gz
Hybrid Assembly¶
Now we are starting to get into the fun stuff! Let's say you have both paired-end Illumina
reads and Nanopore reads for a sample. You can use Bactopia to create a hybrid assembly
using both sets of reads. To do this you will pass the reads using --r1, --r2 (for
Illumina reads), and --ont (for Nanopore reads). Alongside these, you will also provide
the --hybrid parameter will tell Bactopia to create a hybrid assembly by using
Unicycler which assembles the short-reads first
then bridges the gaps with the long-reads.
Use --r1, --r2, --ont, and --hybrid for hybrid assembly
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz \
--ont /path/to/my-sample.fastq.gz \
--hybrid
Hybrid Assembly (Short-read Polishing)¶
Very similar to --hybrid, you will pass the reads using --r1, --r2 (for Illumina reads),
and --ont (for Nanopore reads). Instead this time you will use --short_polish which will
tell Bactopia to create a hybrid assembly using Dragonflye
to assemble the long-reads first then polish with the short-reads.
Use --r1, --r2, --ont, and --short_polish for hybrid assembly with short-read polishing
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz \
--ont /path/to/my-sample.fastq.gz \
--short_polish
Prefer --short_polish over --hybrid with recent ONT sequencing
Using Unicycler (--hybrid) to create a hybrid
assembly works great when you have low-coverage noisy long-reads. However, if you are
using recent ONT sequencing, you likely have high-coverage and using the --short_polish
method is going to yeild better results (and be faster!) than --hybrid.
Well! These are all the ways you can process your local Illumina and/or Nanopore reads. Now, onto assemblies!
Assembly¶
Let's imagine, for what ever reason, you don't have access to the raw reads for a sample,
only the assembly. It happens, but Bactopia has you covered! You can use the --assembly
parameter to tell Bactopia to use the assembly for downstream analyses.
Now when you provide an assembly a few things happen.
- Assemblies will have 2x250bp Illumina reads simulated without insertions or deletions in the sequence and a minimum PHRED score of Q33.
- By default, the input assembly will be used for all downstream analyses (e.g. annotation)
which use an assembly. Otherwise, if the
--reassembleparameter is given, then an assembly will be created from the simulated reads.
Use --assembly for an assembled FASTA
bactopia \
--sample my-sample \
--assembly /path/to/my-sample.fna.gz
ENA/SRA Accession¶
Bactopia's predecessor, Staphopia, relied heavily on the ability to access publicly available FASTQs from the European Nucleotide Archive (ENA) and the Sequence Read Archive (SRA). It was important this ability to rapidly access millions of samples was maintained in Bactopia.
So, if you find yourself wanting to include publicly available samples in your analysis,
Bactopia has that built in for you! You can give a provide an Experiment accession
(--accession), and Bactopia will use fastq-dl to
automatically download associated FASTQ files from either ENA or SRA. Then the downloaded FASTQ
file will be processed by Bactopia just like your normal local FASTQs.
Use --accession to process an Experiment accession
bactopia \
--accession SRX000000
Why only Experiment accessions?
In the grand scheme of accession hierarchies, Experiment accessions are really the only unique ones. For example, a multiple Run accessions can be associated with a single Experiment accession. Or, multiple Exeriment accessions can be associated with a single BioSample accession. So, by using Experiment accessions, you can be confident you are getting only the sequences associated with that "unique" Experiment.
I only have a XYZ accession, what now?
That's not an issue at all! You can make use of bactopia search to quickly find
any Experiment accessions associated with your accession. Please see the examples
below for more information.
What happens when an Experiment has multiple Runs?
In cases where a single Experiment might have multiple Run accessions associated with it, the FASTQ files from each Run are merged into a single set of sequences.
NCBI Assembly Accession¶
If you can process assemblies, and seamlessly download FASTQs from ENA/SRA, it only makes
sense that you could also process assemblies from NCBI Assembly! Similar to downloading
FASTQs from ENA/SRA, you can provide an NCBI Assembly accession using --accession. These
accessions are the ones that start with GCF or GCA. When provided an NCBI Assembly
accession Bactopia will use ncbi-genome-download
to go fetch the associated assembly and process it just like a local assembly.
Use --accession to process an NCBI Assembly accession
bactopia \
--accession GCF_000000000
Do I need to provide the assembly version? (e.g. GCF_000000000.1)
Overtime I've found the assembly version to be unstable. For example, sometimes an assembly might be corrected, and the previous version is not made available any longer. So, to avoid any issues, Bactopia will always use the latest version of a given NCBI Assembly accession.
Multiple Samples¶
By this point you should have a good understanding of how to process a single sample, but you might be thinking: "I have hundreds of samples, I don't want to run Bactopia hundreds of times. Can I just run them all at once?" The answer is YES!
Bactopia allows you to provide a file of filenames (FOFN) using --samples or a list
of accessions using --accessions. Using either of these parameters, allows you to process
a single or thousands of samples
in a single command.
In this section, we will look at the how to process multiple samples using --samples and
--accessions. We will also look Bactopia Helper commands to assist in generating the
appropriate FOFN or accession list to process multiple samples.
Here's a little table to help you decide which parameter to use:
| Parameter | Application | Helper Command |
|---|---|---|
--samples |
Local Samples | bactopia prepare |
--accessions |
ENA/SRA & Assembly Accessions | bactopia search |
Now, let's look into each of these in more detail.
Local Samples¶
Above you learned how you could use parameters like --r1 and --r2 to process a single sample,
but you have a lot of samples you just received sequences for. Now you plan to run Bactopia
on each sample, so let's learn how to generate a FOFN, or samplesheet, you can pass to
Bactopia to process all samples at once.
First, I've thrown out file of filenames (FOFN) a few times now, but what is it? A FOFN, a file that contains a list of samples and their associated FASTQs/FASTAs. A file, of filenames.
For Bactopia this FOFN is a tab-delimited table with five columns:
| Column | Description |
|---|---|
| sample | A unique prefix, or unique name, to be used for naming output files |
| runtype | Informs Bactopia what type of input the sample is (e.g. paired-end, single-end, nanopore, etc...) |
| genome_size | The expected genome size for the given sample |
| species | The expected taxonomic classification for the given sample |
| r1 | If paired-end, the first pair of reads, else the single-end reads |
| r2 | If paired-end, the second pair of reads |
| extra | Either the assembly or long reads associated with a sample |
With this in mind, let's look at an example FOFN:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 Bacterial species /fq/s01_R1_001.fastq.gz /fq/s01_R2_001.fastq.gz
s02 paired-end 180000 Bacterial species /fq/s02_R1_001.fastq.gz /fq/s02_R2_001.fastq.gz
s03 single-end 180000 Bacterial species /fq/s03_001.fastq.gz
With this FOFN, you can use --samples to process all three samples at once.
Use --samples for Multiple Local Samples
Using --samples can turn into a huge time saver for you, and it is always recommended
to take this approach when possible.
bactopia \
--samples my-samples.txt
Now, you might be thinking, "I don't want to create a FOFN by hand, that's a lot of work!"
Well, lucky you! Bactopia has a built in helper command to help you generate a FOFN
automatically. Let's take a look at bactopia prepare.
bactopia prepare¶
While manually creating the necessary FOFN is possible, it's not recommended. It can be a
bit tedious and error-prone, so please avoid manually creating your FOFN. Instead, use
bactopia prepare to help accurately generate a FOFN for your samples.
When Bactopia recieves a FOFN, the first thing Bactopia does is verify all input files are found and compressed using Gzip. If everything checks out, each sample will then be processed, otherwise a list of samples with errors will be output to STDERR.
Use --check_samples to only validate the FOFN
If you would like to only validate your FOFN (and not run the full pipeline), you can use
the --check_samples parameter. However, if you used bactopia prepare to generate your
FOFN it should be valid.
Honestly, bactopia prepare is one of those tools that is best explained by example. So,
let's take a look at a few examples.
Examples¶
Using bactopia prepare can be a bit tricky at first, but once you get the hang of it, you
will find yourself using it all the time.
Use nice file names
bactopia prepare defaults to <SAMPLE_NAME>_R1.fastq.gz and <SAMPLE_NAME>_R2.fastq.gz
for paired-end reads, and <SAMPLE_NAME>.fastq.gz for single-end reads. Using filenames
that following this will help you avoid using regular expressions.
bactopia prepare should be handle your set up to generate the appropriate, but you might
have to work for it. Let's take a look at available parameters and a few examples.
Available bactopia prepare Parameters
| Parameter | Description |
|---|---|
--path |
The directory where your FASTQs/FASTAs are stored. |
--assembly_ext |
The extension of your FASTAs. Default: .fna.gz |
--fastq_ext |
The extension of your FASTQs. Default: .fastq.gz |
--fastq_separator |
The character to split the FASTQ name on. Default: _ |
--pe1_pattern |
The regular expression to match the first pair of paired-end reads. Default: ([Aa]|[Rr]1|1) |
--pe2_pattern |
The regular expression to match the second pair of paired-end reads. Default: ([Bb]|[Rr]2|2) |
--merge |
Flag samples with multiple read sets to be merged by Bactopia. |
--ont |
Flag single-end reads to be treated as Oxford Nanopore reads. |
--recursive |
Flag to recursively search directories for FASTQs/FASTAs. |
--prefix |
Replace the absolute path with a given string. Default: Use absolute path |
--metadata |
Metadata per sample with genome size and species information. |
--genome-size |
Genome size to use for all samples. |
--species |
Species to use for all samples (If available, can be used to determine genome size). |
--taxid |
Use the genome size of the Taxon ID for all samples. |
Illumina Reads
Let's say you have a directory of paired-end Illumina reads. The files are named to match
the default expectations: <SAMPLE_NAME>_R1.fastq.gz, <SAMPLE_NAME>_R2.fastq.gz, and
<SAMPLE_NAME>.fastq.gz. You can use bactopia prepare to generate a FOFN for you.
bactopia prepare --path /path/to/fastqs
This will generate a FOFN that looks like this:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1.fastq.gz /path/to/fastqs/s01_R2.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1.fastq.gz /path/to/fastqs/s02_R2.fastq.gz
s03 single-end 180000 unknown /path/to/fastqs/s03.fastq.gz
Oxford Nanopore Reads
Let's say you have a directory of Oxford Nanopore reads. The files are named to match
the default expectations: <SAMPLE_NAME>.fastq.gz. You can use bactopia prepare to
generate a FOFN for you.
bactopia prepare --path /path/to/fastqs --ont
By using --ont, any single-end reads that are found will be treated as ONT
reads. Using this will generate a FOFN that looks like this:
sample runtype genome_size species r1 r2 extra
s03 ont 180000 unknown /path/to/fastqs/s01.fastq.gz
Illumina Paired-End and Oxford Nanopore Reads
Let's say you have a directory of paired-end Illumina reads and Oxford Nanopore reads.
Again, they are named to match the default expectations: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz, and <SAMPLE_NAME>.fastq.gz. You can use bactopia prepare
to generate a FOFN for you.
bactopia prepare --path /path/to/fastqs --ont
Again, use --ont to tell bactopia prepare to treat any single-end reads as
ONT reads. Using this will generate a FOFN that looks like this:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1.fastq.gz /path/to/fastqs/s01_R2.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1.fastq.gz /path/to/fastqs/s02_R2.fastq.gz
s03 ont 180000 unknown /path/to/fastqs/s03.fastq.gz
Merging Multiple Illumina Runs
Let's say you have a directory of Illumina reads, but you have multiple runs
for each sample and want Bactopia to merge the reads. Again, assuming they
are named to match the default expectations: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz, and <SAMPLE_NAME>.fastq.gz. You can use bactopia prepare
to generate a FOFN for you.
bactopia prepare --path /path/to/fastqs --merge
By using --merge, any samples with multiple runs will be merged into a single set
of reads. Using this will generate a FOFN that looks like this:
sample runtype genome_size species r1 r2 extra
s01 merge-pe 180000 unknown /run1/s01_R1.fastq.gz,/run2/s01_R1.fastq.gz /run1/s01_R2.fastq.gz,/run2/s01_R2.fastq.gz
s02 merge-se 180000 unknown /run1/s02.fastq.gz,/run2/s02.fastq.gz
Reads with '*_001.fastq.gz' names
Let's say you have a directory of Illumina reads, but they are named with
*_001.fastq.gz instead of the default expectations: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz, and <SAMPLE_NAME>.fastq.gz. You can use bactopia prepare
but you will have to provide a few extra parameters to generate a FOFN for you.
bactopia prepare --path /path/to/fastqs --fastq-ext '_001.fastq.gz'
Here you will need to use --fastq-ext to tell bactopia prepare to look for
*_001.fastq.gz instead of the default *.fastq.gz. Using this will generate a
FOFN that looks like this:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1_001.fastq.gz /path/to/fastqs/s01_R2_002.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1_001.fastq.gz /path/to/fastqs/s02_R2_002.fastq.gz
s03 single-end 180000 unknown /path/to/fastqs/s03_001.fastq.gz
There are many possible combinations of parameters you can use with bactopia prepare,
if you have one you are stuck on or would like to see an example of, please let me know
by Submitting and Issue on GitHub.
Accessions¶
If you started from the top, and made it this far I commend you! Eitherway, above you
learned you could use --accession to download FASTQs from ENA/SRA or assemblies from
NCBI Assembly. Then you just learned you could use --samples to process as many samples
as you want. So, it only makes sense that there would be a complement to --samples for
processing multiple accessions at once! This parameter is --accessions.
Use --accessions for Multiple Accessions
Using --accessions can turn into a huge time saver for you, by allowing you to
process as many publicly available genomes as you want.
bactopia \
--accessions my-accessions.txt
Similarly, to --samples, there is a complementary helper command called bactopia search
that will allow you to submit a query and generate a list of Experiment accessions to be
processed by Bactopia (via --accessions).
Let's take a look at bactopia search and how it can help you.
bactopia search¶
bactopia search has been made to help assist in generating a list of Experiment accessions
to be procesed by Bactopia (via --accessions). You can provide a Taxon ID (e.g. 1280), a
organism name (e.g. Staphylococcus aureus), a Study accession (e.g. PRJNA480016), a BioSample
accession (e.g. SAMN01737350), or a Run accession (e.g. SRR578340). This value is then
queried against ENA's Data Warehouse API),
and a list of all Experiment accessions associated with the query is returned.
Again, it's probably easier if we just look at a few examples.
Examples¶
First we'll look at a single example in order to provide a description of the output files.
bactopia search --query PRJNA480016 --limit 5
INFO 2023 00:root:INFO - Submitting query (type - bioproject_accession) search.py:472
INFO 2023 00:root:INFO - Writing results to ./bactopia-metadata.txt search.py:554
INFO 2023 00:root:INFO - Writing accessions to ./bactopia-accessions.txt search.py:564
INFO 2023 00:root:INFO - Writing filtered accessions to ./bactopia-filtered.txt search.py:569
INFO 2023 00:root:INFO - Writing summary to ./bactopia-search.txt search.py:575
In the above command we are searching for all Experiment accessions associated with the
Study accession PRJNA480016. However, the --limit parameter is used to limit results
to just 5 Experiment accessions. Then multiple files are produced:
| Extension | Description |
|---|---|
-metadata.txt |
A tab-delimted file of all results from the query |
-accessions.txt |
A list of Experiment accessions to be processed |
-filtered.txt |
A list of any Experiment accessions that were filtered out, otherwise an empty |
-search.txt |
A summary of the completed request |
Example bactopia-metadata.txt
When completed a file called bactopia-metadata.txt is produced. This file contains
multiple fields (sample_accession, tax_id, sample_alias, center_name, etc...) for
each Experiment accession returned by the query.
run_accession project_name submission_accession library_selection last_updated sra_bytes collected_by isolate fastq_bytes instrument_platform sra_aspera fastq_galaxy country sample_description experiment_title sra_galaxy fastq_md5 sample_accession secondary_study_accession read_count study_title collection_date_end sample_title instrument_model description sra_md5 fastq_ftp base_count library_strategy location library_source sra_ftp library_layout location_start status lon fastq_aspera host_sex sample_alias collection_date_start run_alias collection_date experiment_alias center_name host library_name tag first_created lat strain experiment_accession scientific_name tax_id study_accession host_scientific_name accession secondary_sample_accession location_end first_public study_alias isolation_source
SRR7706353 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 595393300 353569630;334112090 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/003/SRR7706353 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz USA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/003/SRR7706353 6cf7a954abc803c8be6515545b321e2d;f879b1fa058e80fa764beb8e333877ae SAMN09847868 SRP158268 1493115 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2023-01-07 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 4f7c2a8836ce2471fec07128f2c9b407 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz 897918508 WGS GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/003/SRR7706353 PAIRED public fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz JE2 2023-01-07 JE2_R1.fastq.gz 2017-07-01 JE2 SUB4273132 Homo sapiens JE2 ena;pathogen;bacterium;datahub;priority 2018-08-18 JE2 SRX4563690 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706353 SRS3680044 2018-08-18 PRJNA480016
SRR7706354 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 227970617 Emory Cystic Fibrosis Biospecimen Registry (CFBR) replicate of CFBRSa66A 129917564;131945147 ILLUMINAfasp.sra.ebi.ac.uk:/vol1/srr/SRR770/004/SRR7706354 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz USA: Atlanta, GA MRSA Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/004/SRR7706354 371165d54adfd1300c7b02e79d8d4245;5517e629b8e8ad00dbd6ef9a5f8d073d SAMN09847839 SRP158268 535939 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2018-01-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 323e7336212b256ba2509a14bd90790a ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz 322122209 WGS 33.749 N 84.388 W GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/004/SRR7706354 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz male CFBRSa66B 2018-01-02 CFBRSa66B_R2.fastq.gz 2012-07-16 CFBRSa66B SUB4273132 Homo sapiens CFBRSa66B ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-150 SRX4563689 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706354 SRS3680043 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706356 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 328642242 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 191742121;188439990 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/006/SRR7706356 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz USA: Atlanta, GA MRSA Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/006/SRR7706356 2b0c01434a7e677c6697ff49985de0f7;08c4f37d7fdbeac0133819ee3af6dd21 SAMN09847834 SRP158268 780838 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2017-09-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ec8397df7897777d1c332522c6227458 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz 469342822 WGS 33.749 N 84.388 W GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/006/SRR7706356 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz male CFBRSa25 2017-09-01 CFBRSa25_R2.fastq.gz 2012-03-26 CFBRSa25 SUB4273132 Homo sapiens CFBRSa25 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-134 SRX4563687 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706356 SRS3680041 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706361 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 599269367 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 353160072;336993031 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/001/SRR7706361 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz USA: Atlanta, GA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/001/SRR7706361 45fa5f0ed629d81282f1429b42c18432;c9c1b6be39fceab54d20d41450776050 SAMN09847850 SRP158268 1496420 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2018-04-04 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 085cbb8f7b186d3f61bab022323f61ce ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz 899864766 WGS 33.749 N 84.388 GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/001/SRR7706361 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz male CFBRSa07 2018-04-03 CFBRSa07_R1.fastq.gz 2012-10-03 CFBRSa07 SUB4273132 Homo sapiens CFBRSa07 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-238 SRX4563682 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706361 SRS3680035 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706362 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 241721284 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 139499004;138853939 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/002/SRR7706362 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz USA: Atlanta, GA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/002/SRR7706362 d6f7434e83969245df356e0c3aaa72e8;4bfa95c3a9db9d93b65b39530f5be0c7 SAMN09847844 SRP158268 572961 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2017-11-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 189012bcc94d59002369ebc17ad303fa ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz 344400515 WGS 33.749 N 84.388 GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/002/SRR7706362 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz male CFBRSa06 2017-11-02 CFBRSa06_R1.fastq.gz 2012-05-16 CFBRSa06 SUB4273132 Homo sapiens CFBRSa06 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-172 SRX4563681 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706362 SRS3680034 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
Example bactopia-summary.txt
When completed a file called bactopia-summary.txt is produced, that will contain a
basic summary of the query results.
Bactopia Summary Report
Total Samples: 1
Passed: 1
Gold: 0
Silver: 1
Bronze: 0
Excluded: 0
Failed Cutoff: 0
QC Failure: 0
Reports:
Full Report (txt): ./bactopia-report.tsv
Exclusion: ./bactopia-exclude.tsv
Summary: ./bactopia-summary.txt
Rank Cutoffs:
Gold:
Coverage >= 100x
Quality >= Q30
Read Length >= 95bp
Total Contigs < 100
Silver:
Coverage >= 50x
Quality >= Q20
Read Length >= 75bp
Total Contigs < 200
Bronze:
Coverage >= 20x
Quality >= Q12
Read Length >= 49bp
Total Contigs < 500
Assembly Length Exclusions:
Minimum: None
Maximum: None
From the output files, you will want to use the file with the -accessions.txt extension. In
the above query the -accessions.txt file looked like this:
SRX4563681 illumina Staphylococcus aureus 2800000
SRX4563689 illumina Staphylococcus aureus 2800000
SRX4563687 illumina Staphylococcus aureus 2800000
SRX4563682 illumina Staphylococcus aureus 2800000
SRX4563690 illumina Staphylococcus aureus 2800000
Use the file with the -accessions.txt extension with --accessions
The file with the -accessions.txt extension is the file you will use with --accessions
to process the Experiment accessions with Bactopia.
Additional Helpful Parameters¶
-profile¶
Bactopia makes use of Nextflow Config Profiles
to specify the executor to use. By default, Bactopia will use the conda profile. There
are other built in profiles including: docker, singularity, slurm, etc... To use a
specific profile, you can use the -profile parameter.
For example if you want Nextflow to use Docker, you would use the following command:
bactopia ... -profile docker
With this, Nextflow will use Docker to run all the processes in Bactopia (even though Bactopia is installed with Conda!).
Always prefer containers over Conda
While I will be the first to admit that I love Conda, it is not perfect. Overtime tools can become broken or incompatible due to dependencies. Containers are a great way to avoid these issues. If you are using Bactopia, and have Docker or Singularity available I would recommend using them over Conda.
-resume¶
Bactopia relies on Nextflow's Resume Feature
to resume runs. You can tell Bactopia to resume by adding -resume to your command line.
When -resume is used, Nextflow will review the cache and determine if the previous run
is resumable. If the previous run is not resumable, execution will start at the beginning.
--max_cpus¶
At execution, Nextflow creates a queue and the number of slots in the queue is determined
by the total number of cores on the system. So if you have a 24-core system, that means
Nextflow will have a queue with 24-slots available. This feature kind of makes --max_cpus
a little misleading. Typically when you give --max_cpus you are saying "use this amount
of cpus". But that is not the case for Nextflow and Bactopia. When you use --max_cpus
what you are actually saying is "for any particular task, use this amount of slots".
Commands within a task processors will use the amount specified by --max_cpus.
--max_cpus can have a significant effect on the efficiency of Bactopia
For example if you have a system with 24-cores.
This command, bactopia ... --max_cpus 24, says for any particular task, use 24 slots.
Nextflow will give tasks in Bactopia 24 slots out of 24 available (24-core machine). In
other words the queue can one have one task running at once because each task occupies
24 slots.
On the other hand, bactopia ... --max_cpus 4 says for any particular task, use 4 slots.
Now, for Nextflow will give each task 4 slots out of 24 slots. Which means 6 tasks can be
running at once. This can lead to much better efficiency because less jobs are stuck
waiting in line.
There are some tasks in Bactopia that will only ever use a single slot because they are
single-core tasks. While others will always use the number of slots specified by
--max_cpus.
If the --max_cpus is too high, you will likely reduce the efficiency of Bactopia.
When in doubt --max_cpus 4 is a safe value.
This is also the default value for Bactopia.
-qs¶
The -qs parameter is short for queue size. As described above for --max_cpus, the
default value for -qs is set to the total number of cores on the system. This parameter
allows you to adjust the maximum number of cores Nextflow can use at any given moment.
-qs allows you to play nicely on shared resources
From the example above, if you have a system with 24-cores. The default queue size if 24 slots.
bactopia ... --max_cpus 4 says for any particular task, use a maximum of 4 slots.
Nextflow will give each task 4 slots out of 24 slots. But there might be other people
also using the server.
bactopia ... --max_cpus 4 -qs 12 says for any particular task, use a maximum of 4
slots, but don't use more than 12 slots. Nextflow will give each task 4 slots out of
12 slots. Now instead of using all the cores on the server, the maximum that can be
used in 12.
-qs might need adjusting for job schedulers.
The default value for -qs is set to 100 when using a job scheduler (e.g. SLURM,
AWS Batch). There may be times when you need adjust this to meet your needs. For
example, if using AWS Batch you might want to increase the value to have more jobs
processed at once (e.g. 100 vs 500).
--genome_size¶
Throughout the Bactopia workflow a genome size is used for various tasks. By default, a genome size is set to 0, and things such as coverage reduction are skipped. However, if you provide an expected genome size, these steps will be enabled.
Use --genome_size to improve results and speed
By providing a genome size, Bactopia will reduce the coverage to a maximum of 100x (default). In doing so, for samples with greater than 100x coverage, you will see a reduction in execution time as well is improved results. This is because, with excessive coverage some tools will produce poorer results while taking much longer.
--nfconfig¶
A key feature of Nextflow is you can provide your own config files. What this boils down to
you can easily set Bactopia to run on your environment. With --nfconfig you can tell
Bactopia to import your config file.
--nfconfig has been set up so that it is the last config file to be loaded by Nextflow.
This means that if your config file contains variables (e.g. params or profiles) already
set they will be overwritten by your values.
Nextflow goes into great details on how to create configuration files. Please check the following links for adjustments you be interested in making.
| Scope | Description |
|---|---|
| env | Set any environment variables that might be required |
| params | Change the default values of command line arguments |
| process | Adjust perprocess configurations such as containers, conda envs, or resource usage |
| profile | Create predefined profiles for your Executor |
There are many other scopes that you might be interested in checking out.
You are most like going to want to create a custom profile. By doing so you can specify it
at runtime (-profile myProfile) and Nextflow will be excuted based on that profile. Often
times your custom profile will include information on the executor (queues, allocations, paths, etc...).
If you need help please reach out!
If you're using the standard profile (did not specify -profile 'xyz') this might not be necessary.
--cleanup_workdir¶
After you run Bactopia, you will notice a directory called work. This directory is where
Nextflow runs all the processes and stores the intermediate files. After a process completes
successfully, the appropriate results are pulled out and placed in the sample's result folder.
The work directory can grow very large very quickly! Please keep this in mind when using
Bactopia (and other Nextflow pipelines). To help prevent the build up of the work
directory you can use --cleanup_workdir to automatically delete the work directory after
a successful run.